The risk analysis (RA) evaluates, controls, mitigates, communicates, and reviews any vulnerability and
risk to improve business operations, considering:
a) Identify and document all possible and potential vulnerabilities, failures, disasters, root causes, and
actions are taken for each business operation, department, or area.
b) Based on the potential vulnerabilities, assign the corresponding risks, and evaluate how these risks should be
minimized as much as possible to an acceptable level.
c) Establish the testing requirements with the associated documentation based on risk, complexity, and novelty
as part of a scalable approach that enables to establish and standardize the selection of the appropriate life cycle
testing or validation activities for a specific business operation.
HOW TO MAKE A RISK ANALYSIS IN (7) STEPS?
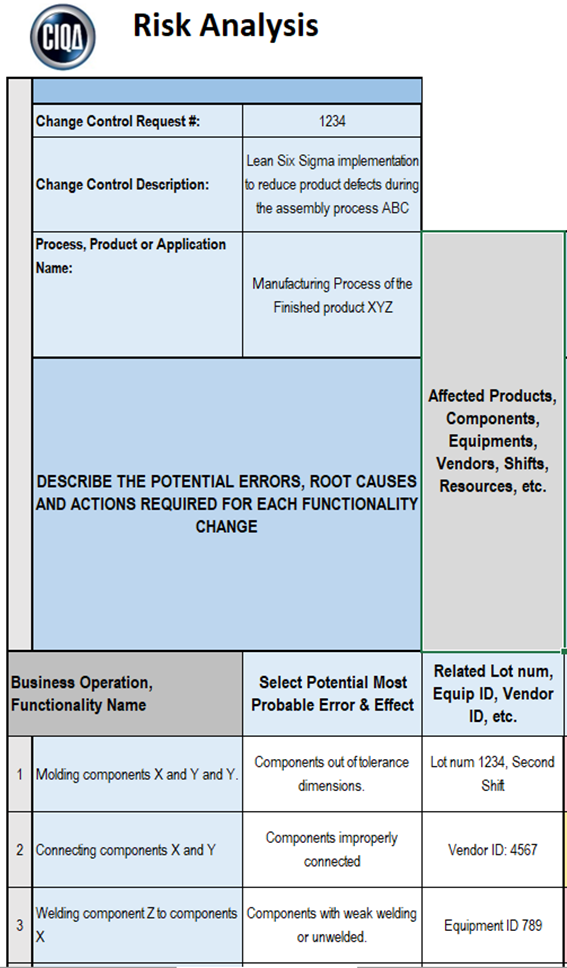
STEP 1 – The first step to create a risk assessment is to define the business operations or function name.
Define the main activities that produce the process quality characteristics and attributes related to each business operation.
This information can be obtained from the URS and assessing how each of the requirements correlates to a similar product
or system function (grouping) in the FRS.
They must be grouped by product, equipment, systems, areas, etc.
For example the manufacturing process of the product ABC consists of three operations:
Molding components X and Y,
Connecting components X and Y
Welding component Z to components X and Y.
As required, perform the same risk analysis for each business operation or activity described in the functional items listed in the URS, manufacturing instruction, test script, etc.
STEP 2 – The second step is to define and describe the potential most probable errors, disruptive incident, or failure effect description.
Add all potential disruptive incidents or effects.
Select the potential most probable error and effect.
For example, the manufacturing process of product ABC presented some typical failures as:
Components out of tolerance dimensions.
Components improperly connected
Components with weak welding or unwelded.
More examples of potential failures and errors:
Other typical and potential errors and effect in computerized systems are:
Functionality Errors
Description
Computational Error
Improper computation of the formulae / improper business validations in code.
Data error
Incorrect data population / update in database
Database Error
Error in the database schema/Design
Missing Design
Design features/approach missed/not documented in the design document and hence does not correspond to requirements
Inadequate/sub-optimal Design
Design features/approach needs additional inputs for it to be complete. Design features described does not provide the best approach (optimal approach) towards the solution required
Incorrect Design
Wrong or inaccurate Design
Ambiguous Design
The design feature/approach is not clear to the reviewer. Also includes ambiguous use of words or unclear design features.
Boundary Conditions Neglected
Boundary conditions not addressed/incorrect
Interface Error
Internal or external to application interfacing error, Incorrect handling of passing parameters, Incorrect alignment, incorrect/misplaced fields/objects, unfriendly window/screen positions
Logic Error
Missing or Inadequate or irrelevant or ambiguous functionality in source code
Message Error
Inadequate/ incorrect/ misleading or missing error messages in source code
Navigation Error
Navigation not coded correctly in source code
Performance Error
An error related to performance/optimality of the code
Missing Requirements
Implicit/Explicit requirements are missed/not documented during the requirement phase
Inadequate Requirements
The requirement needs additional inputs to be complete
Incorrect Requirements
Wrong or inaccurate requirements
Ambiguous Requirements
The requirement is not clear to the reviewer. Also includes ambiguous use of words, e.g. Like, such as, maybe, could be, etc.
Sequencing / Timing Error
Error due to incorrect/missing consideration to timeouts and improper/missing sequencing in source code.
Incorrect Standards Use
Standards did not follow like improper exception handling, project-related design/requirements/coding standards
System Error
Hardware and Operating System-related error, Memory leak
Test Plan / Cases Error
Inadequate/ incorrect/ ambiguous or duplicate or missing-Test Cases & Test Scripts, Incorrect/Incomplete test setup
Typographical Error
Spelling / Grammar mistake in documents/source code
Variable Declaration Error
Improper declaration/usage of variables, Type mismatch error in source code
STEP 3 – The third step is to define the affected products, components, equipment, vendors, shifts, resources, clients, suppliers, etc.
For the example of the manufacturing process of product ABC, some possible related failure factors should be:
The second shift, Lot number 1234
Component supplier/vendor ID 4567
Welding machine: 789
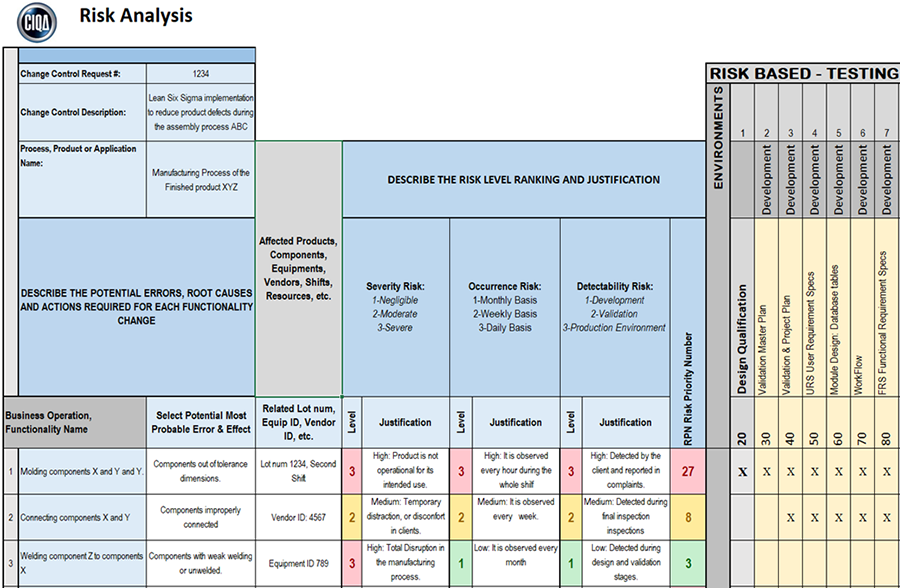
STEP 4 – The fourth step is to define the severity risk value.
Determine the risk dangerousness associated as HIGH, MEDIUM or LOW for each business operation or functionality name (e.g. URS, FRS, etc) in terms of potential failure or if a particular product, equipment, or system attribute or characteristic is not available (either partially, complete, off-line, non-functioning, intermittent, etc.).
Below the severity levels are discussed and listed in order of escalation.
Risk Level Number
Risk Severity Category
Description
1
Negligible, Minor, Low
Limited to no product malfunction, business disruptions, or property damage.
More information
Minor products, equipment, or application functionality’s fault or defects are tricky to classify. While they still should not impede the primary features or functionality. This functionality’s failure can cause or has the potential to cause slight discomfort, inconvenience, or dissatisfaction in some customer(s). They detected only a few light problems in the appearance and performance of the functionality, etc. The functionality is not affecting or annoying significantly the client or administrator by such a minor fault. The clients never claim or complaint about these errors or failure, nor refuse or hold to use the product, equipment o application such as; but not limited to:
– Field Name and Input Character (Size)
– Menu Options, name, location, or size change
– Non-Configurable Standardized Field
-Terms and Conditions
– Appearance settings, screen colors, etc.
– slow system response, high latency.
– no system information messages are displayed,
– no error messages are displayed after some abnormal situation.
– no helping tool, wizard, or intuitive indicative menus or navigation options to reach and accelerate the user access, operations, and needs in the application.
– no reporting features satisfying the user expectative for data evaluation and summarizing.
– no main core operation functionalities: calendar, chat, support, online documentation.
2
Medium, Marginal or moderate
A hindrance that may affect business operations without shutting down, you have no minor damage, it may be an occurrence in the surrounding neighborhood. Temporary disruptions of business or major damage to the facility, impacts are to the community, clients, and employees.
More information:
Product, equipment, or application functionality’s fault can generate some distraction, dissatisfaction, or discomfort in customers, administrators, and users. Clients feel annoying, but, most of the time do not make a formal complaint, clients do not refuse to use the application since it can be used with this potential failure, defect or fault however do not feel 100% satisfied with the situation, such as; but not limited to anomalies in:
– Advertisement functionalities
– Automatic Training Verification functionalities
– Calendar functionalities
– Configurable Standardized Field functionalities
– Dashboard functionalities
– Delegation of Account functionalities
– Mail functionalities
– Merge functionalities
– Messenger functionalities
– Notes functionalities
– Notifications functionalities
3
Catastrophic, High
An event that affects the entire regional community causing business disruptions and forces closure of building(s). This is an event of large proportions. It can include complete destruction, multiple injuries or deaths, and a regional event which means limited or no outside resources available for prolonged periods of time.
More information:
Business operation containing a severe potential risk failure with a high degree of customer dissatisfaction. The product, equipment, or application functionality is NOT operational for its intended use, produce not consistent results, produce not accurate output(s), can hold it operation, it can stop, or it can be rejected by the client, promote that the client(s) will refuse to use it; and can promote a customer claim or complaint, potential loss of client(s) for dissatisfaction. It may represent a serious business loss of confidence, credibility, trust, market, clients, money, confidential, privileged, or personal information, potential failures related to fraud that may affect: payments done and received, including but not limited to anomalies in functionalities such as:
– Data integrity, transactions, and calculations functionalities.
– Import and export data functionalities
– Data depository: upload and download information functionalities
– System Security, Log-In & Log-Out, and Access Controls to confidential information.
– Electronic signatures and electronic record functionalities
– Decision making and control logic functionalities
– Backup and restore capabilities and functionalities
– Audit Trail (History Log) functionalities
– Crosslink/ Relationship functionalities
– Mayor changes in forms, menus, pushbuttons, and reports functionalities
– High Availability (HA) functionalities and others.
These are issues that prevent a business from meeting requirements or carrying out a feature.
Critical bugs, or showstoppers as they are often called, are so severe that they prevent from further testing. Critical issues manifest themselves in a number of different ways and can range from an app that continuously crashes, to a button missing in the user interface that prevents you from loading or triggering a required part of the application.
Add a justification for your selection of severity risk level ranking.
Step 5 – The fifth step is to define the occurrence risk value.
Determine how often the failure or error is observed.
Establish the likelihood of occurrence as HIGH, MEDIUM, or LOW for each potential error or failure observed and associated with the business operation or functionality name (e.g. URS, FRS, etc)
The highest-ranking goes to the event that can be expected to occur at some point, with lower rankings to those events that only might occur, or are not expected to occur at all.
Risk Level Number
Occurrence Category
Definition
3
High
A failure is or may be observable very frequently, many occurrences, every time the user utilizes the application at least one (1) or more failures per date. This kind of failure render a consistent and reproducible error every time the business operation or activity is done every minute, hour, or daily basis.
2
Moderate
A failure is or may be observable occasionally, the frequency of this type of error is on a weekly basis (faults that do not occur daily but occur weekly). In addition, these failures can be of intermittent occurrence, not consistently or frequently observable at least weekly, but not necessarily on a daily basis.
1
Low
A failure is or may be observable in very few occasions, with maximum occurrence once a month. Most of the time it takes more than one (1) month to be observable or return it to occur. In addition, these failures can be of intermittent occurrence, not consistently or frequently observable on a monthly basis, but not necessarily on a weekly basis.
Add a justification for your selection of occurrence risk level ranking.
Step 6 – The sixth step is to define the detectability risk value.
Risk Level Number
Detectability Category
Definition
1
High
Potential functionality’s fault is detected most of the time at its origin, source, or in the first step or stage of inspection of the development testing process either dry runs or commissioned by the designers or initial testing representative. Very easily identifiable fault or detectable very quickly in the process through simple inspection or intrinsic mechanisms of the process of development, it can be frequently detected in the development or implementation environment.
2
Moderate
Potential functionality’s fault not detected typically in the first stage of inspection in the development or implementation environment, but can be detected on second inspections or subsequent inspections and testing performed as part of the validation, verification, and testing processes on the testing environment. These failures are detected within the company before the product or service is released to the customer.
3
Low
Potential functionality’s error or fault with a low probability to be detected by the primary and secondary inspections stages and testing process within the company at the development and testing environment(s). These types of faults are more probable to be detected by the clients once the product or service is released, distributed, and used by many & different clients, roles/positions under the production environment(s). Most of the time, the failure is detected by the client, customer or an end-user or administrator under simulation testing in a production environment.
Add a justification for your selection of detectability risk level ranking.
STEP 7 – The seventh step is to calculate the RPN risk value.
Determine the risk priority number (RPN) for each business operation, functionality and its associated potential failure or error(s).
This value is obtained by multiplying three (3) risk factors described before:
RPN = Severity Risk Number x Occurrence Risk Number x Detectability Risk Number
The best practices approach developed below is a color code of a three-level system with low (green) medium (yellow) and high (red) risk categories.
These can be characterized as follows:
High Risk: Failure would severely affect safety and quality processes.
Medium Risk: Failure would have a moderate impact on safety and quality processes.
Low Risk: Failure would have a minor impact on patient safety or product quality.
From this definition, a standard risk matrix* has been built, for example:
* Sample matrix – your organization MUST develop (and justify) your own criteria. Where the horizontal columns are for safety/severity/quality, and the vertical rows represent probability/frequency/detectability. Utilize the acceptance criteria chart to find overall risk.
Place the overall risk into one of the three risk severity classes (High/Medium/Low).
After developing the acceptance criteria the process proceeds as follows to document a risk assessment as shown in the following figure:
IN-TOLERABLE REGION
Unacceptable – Catastrophic Risk
TOLERABLE REGION
As Low As Reasonably Practical – ALARP Risk
ACCEPTABLE REGION
Harmless Risk
Risk management tool
Risk Mitigation Actions
Based on the risk region obtained after calculating the RPN, risk mitigation is often accomplished by the introduction of a series of steps or processes that lower the likelihood of unsafe use.
– May reinforce good clinical practices
– May introduce new risk mitigation measures
– May include administrative checks to support risk mitigation efforts
Each intervention that is part of risk mitigation may introduce some level of burden
Mitigation activities and priority level criteria are then applied to the functional items (FRS) and the output is considered in the validation plan, IQ, OQ, and PQ test scripts, as described in the section below.
Risk-Based Validation Approach – Priority Level
A risk-based validation approach for each functional category can be established once the risk assessments for individual functional items from the URS have been determined.
The following best practice approach outlines three types of validations that can be utilized with a risk-based process.
High: Complete/comprehensive testing required. All systems and sub-systems must be thoroughly tested according to a scientific, data-driven rationale. This is similar to the ‘classic’ approach to validation. Furthermore, enhancement may be needed to improve the detectability of failure via in-process production controls.
Medium: testing of the functional requirements per the URS/FRS required with sufficient assurance that the item has been properly characterized.
Low: No formal testing needed, but the presence (detectability) of the functional item may be required.
Hazard Analysis
Determination of the mitigation actions, testing, and validation efforts based on the risk region:
Based on the calculated RPN risk priority number, determine the mitigation actions based on risk region using color code.
Risk Region Category
Risk Region Description
Risk Mitigation Actions, testing and validation requirements recommended, as applicable.
Unacceptable Region
This intolerable risk is identified in region color red since denotes the highest risk obtained considering the combined factors of severity, occurrence, and detectability. It must require intervention to re-evaluate and revise the design to improve it, considering preventive and corrective actions. The corresponding application functionalities will be classified as intolerable functionalities with potential permanent impairment. Complete/comprehensive testing required.
High: Re-evaluate design specifications and requirements, product hold, Recall or reject, CAPA investigation, Full validation, change control management and retraining required, 100% inspection, high sampling plan recommended, SCAR, FDA notification, Full audit, etc.
Tolerable Region
This medium risk region is identified in the color yellow, since denotes “as low as reasonably practical” risk (ALARP) obtained considering the combined factors of severity, occurrence, and detectability. It only suggests to re-evaluate the design to improve it considering preventive and corrective actions, but, it is not necessary as mandatory actions, since they are classified as practical functionalities.
Medium: Non-conformance investigation, evaluate trending, and process variations to prevent any potential intolerable risk. Normal Sampling plan recommended, Fractional Validation and audits required, etc
Acceptable Region
This risk region is identified in color green since denotes negligible or minimum effects with the lowest risk obtained considering the combined factors of severity, occurrence, and detectability. It does not require to re-evaluate the design to improve it considering preventive and corrective actions since they are classified as harmless functionalities. No formal testing needed.
Low: Minimum validation and audit required, existing process monitoring shall be sufficient, reduced sampling plan, not necessary to evaluate design specifications or requirements, etc.
The Risk Priority obtained as (RPN) helps to focus attention on areas where the product or system functionality and client is most exposed to hazards. In order to use resources most effectively, functional risk analysis should be focused on functions with the highest potential impact. Refer to the following example figure.
Risk based testing
The risk-based validation approach can be established also using different risk assessment formats, such as FMEA.
Want to learn more about the
QUALITY RISK ANALYSIS?
Subscribe and follow us on social media.
More details on specific FDA expectations for risk analysis can be found in the guidance document below.
Three (3) options to create a risk analysis or protocol.
Option 1. You can create a great protocol, using a template.
You can download a free sample of a validation template in .pdf format.
To see the complete list of the most popular risk analysis and validation templates, click here.
In addition, you can request a quotation to buy online a full risk management template document in MS Word format that is completely editable, ready to fill and adapt to your needs.
Option 2. We can bring you a formal training on how to create your own risk analysis using our template(s).
This option is recommended if you want to learn more about how to build a robust risk analysis protocol. One of our expert(s) will provide online step-by-step training to your team (unlimited assistance) on how to build reliable risk management using a template. You can improve your corporate validation procedures and policies incorporating our template sections. It includes the template, an exam, and a training certificate for each assistant. Request a quotenow.
Option 3. We can create a customized risk analysis for your company.
One of our expert(s) will create and prepare for you a customized risk analysis procedure with the inputs and specific information of your company. It may include, online support in document creation, execution, or final reporting, Request a quote online.
A required risk management plan that uses risk mitigation strategies beyond FDA‐approved FDA professional labeling.
FDA Amendments Act of 2007 authorized the FDA to require sponsors to develop and comply with REMS programs if determined necessary to ensure the benefits outweigh the risks. – FDA does not directly regulate healthcare professionals or patients who may be impacted by a REMS.
Applies to NDAs, BLAs, and ANDAs. • REMS can be required pre‐ or post‐approval.
Designed to achieve specific goals to mitigate risks associated with the use of a drug.
FDA specifies the required elements of a REMS.
Drug sponsors develop the REMS program based on the required elements. FDA reviews and approves the REMS.
Each REMS has specific safety measures that are targeted to the serious risk(s) associated with the drug or class of drugs.
All REMS include elements, communication, and/or educational materials to communicate risk information to various stakeholders.
More details on specific risk management systems can be found in:
CIQA President and CEO.
I've been working in validation engineering since 1992 with many multinational pharmaceutical companies. I love sharing my passion and knowledge with others. If you have any questions about anything (or just have general questions). I will be more than happy to assist you. You can count on the BEST customer service on CIQA. I go to great lengths to make sure my clients are 100% satisfied with their purchases and check emails/messages consistently throughout the day. You can rest assured that everything being sold here is as-described or your money back. I look forward to working with you!